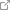ВЕТЕРИНАРНЫМ СПЕЦИАЛИСТАМ.
Задать вопрос президенту НАВНУ

к.в.н. Роман-А. Леонард, практикующий ветеринарный врач, руководитель Центра ветеринарной нефрологии и урологии, президент Российской Научно-практической Ассоциации Ветеринарных Нефрологов и Урологов (НАВНУ)
E-mail: vetnefro@mail.ru
Повреждающие (эффекторные) механизмы хронических гломерулярных и тубуло-интерстициальных заболеваний у собак и кошек и факторы местной самозащиты почек
Введение
На сегодняшний день изучено большое количество различных эффекторных механизмов, принимающих участие в инициации и поддержании процессов клеточного и тканевого повреждения и воспаления при различных хронических асептических заболеваниях почек у животных. Эти повреждающие механизмы можно разделить на первичные и вторичные (таблица 1).
Вместе с тем, ткани и форменные элементы почки имеют в своем арсенале большое количество локальных защитных медиаторных систем и механизмов, способных нивелировать негативное воздействие повреждающих факторов. Работа этих систем местной самозащиты почки является такой же генетически запрограммированной реакцией, как и иммунный ответ при инфекционных заболеваниях. И интенсивность развития хронических нефропатий, и исход заболевания во многом зависят от способности почечного «иммунитета» противостоять разнообразным и разнонаправленным повреждающим факторам.
Уровень кровяного давления в клубочке в норме поддерживают две системы – сосудосуживающая (вазоконстрикционная) и сосудорасширяющая (вазодилатирующая). В норме работа этих систем уравновешена и является неотъемлемой частью поддержания гомеостаза организма. Однако при патологии в силу различных причин (повреждение фенистированного эндотелия и других участков фильтрационного барьера, потеря общей массы функционирующих нефронов (1-4), процессы склерозирования в петлях клубочка и т.д.) это хрупкое равновесие смещается в сторону гиперактивации сосудосуживающего звена и является причиной патологической гиперфильтрации в клубочке, протеинурии и гломеруло- и нефросклероза.
Важным является и то обстоятельство, что одни и те же биологически активные вещества (БАВ): АГ II (схема 1), эндотелин-1, медиаторы продуцируемые клетками воспалительного инфильтрата (фото 1) и т.д. в зависимости от концентрации и взаимодействия с разнотипными рецепторами клеток почечной паренхимы могут быть либо звеном в патогенезе нефропатии, либо являться фактором местной самозащиты.
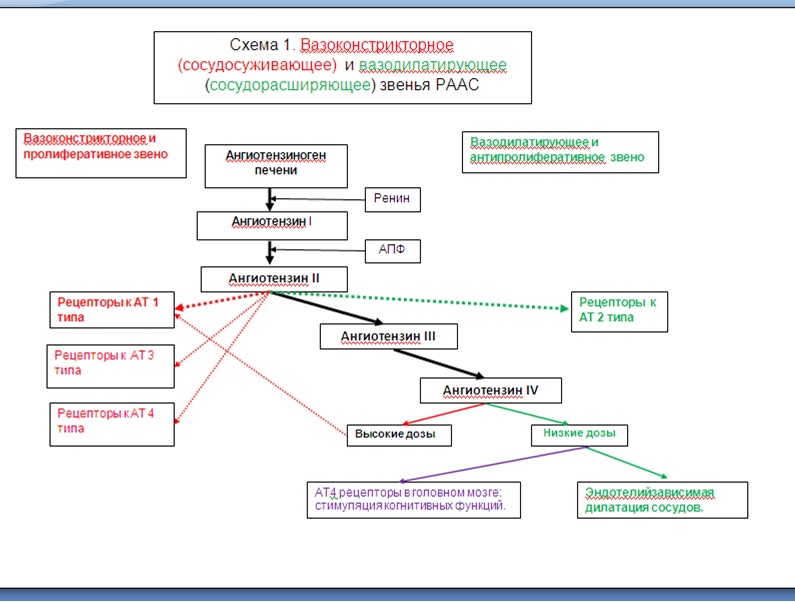
Комментарий к схеме 1.
После открытия РААС в 1950-х годах длительное время считалось, что ее функции сводятся к сужению сосудов (в т.ч. почечных артериол), стимуляции выработки альдостерона, задержке натрия и жидкости в организме, стимуляции жажды и повышению кровяного давления в целом. Однако позже было установлено, что каскад превращений ангиотензиногена печени не прерывается на ангиотензине II (АТ II). Также были открыты рецепторы к АТ II второго типа, воздействие на которые имеет диаметрально противоположный эффект тому, который реализуется через рецепторы первого типа к этому БАВ. И если в норме роль рецепторов второго типа к АГ II не велика (в силу их значительно меньшего количества, чем рецепторов первого типа), то при патологии эффекты, реализуемые через них, могут вносить важный вклад в работу вазодилятируещего звена РААС.
Механизмы иммуноопосредованного повреждения клубочков и факторы тканевой самозащиты почки при ГП и ГН
Начальным этапом в патогенезе как острых, так и хронических гломерулопатий (ГП) и гломерулонефритов (ГН) является повреждение фильтрационного барьера (ФБ) микрокапиллярной сети клубочков. Причиной этих повреждений может быть спадание и слипание капиллярных петель при преренальной почечной недостаточности, повреждение фильтрационного барьера по средствам различных иммуноопосредованных механизмов, а также являться следствием генетически наследуемых или вторичных нефропатий (таблица 2).
Иммунный ответ, приводящий в конечном итоге к возникновению ГП и ГН, реализуется сложной многокомпонентной системой, включающей в себя:
- звенья клеточного и гуморального иммунитета (особенно системы комплемента);
- сигнальные молекулы (тканевой фактор роста TGF-β (5), провоспалительные цитокины и т.д.(6, 11-13));
- тромбоциты и другие компоненты системы свертывания крови.
Большое влияние на интенсивность этого процесса имеет выраженность пери- и интрагломерулярной , перитубулярной и тубулоинтерстициальной инфильтрации агранулоцитами , вазоконстрикция микрокапиллярной сети почки в результате дефицита оксида азота , в норме вырабатываемого эндотелием сосудов и накопление в месте воспаления метаболитов арахидоновой кислоты и кислородных радикалов.
Эффекторным (повреждающим) звеньям патогенеза (таблица 3) при ГП и ГН противостоит система местной индуцированной самозащиты почки. И фатальность патологических изменений в ее тканях зависит от баланса локальных повреждающих факторов и нивелирующих их медиаторов самозащиты или, так называемого, «противовоспалительного статуса» почки (таблица 4). Факторы самозащиты почки могут секретироваться как клетками воспалительного инфильтрата, так и резидентными клетками почки (в первую очередь мезангиальным матриксом). Причем именно в «дефектах» работы местной противовоспалительной системы почки и следует искать первопричину возникновения и развития различных ГП, ГН и тубулоинтерстициальных поражений почек, как у животных, так и у человека. А разработке терапевтических тактик, способных нормализовать работу местной самозащиты почек на доклиническом и, что особенно важно, азотемическом этапах почечного континуума (когда эффективность стандартной нефропротективной терапии не высока и уменьшается параллельно снижению СКФ), в последнее время придается ключевое значение.
|
Таблица 4. Противовоспалительные факторы тканей почки (28). |
|
|
Внеклеточные (локализующиеся на поверхности клеток). |
|
|
Ингибиторы цитокинов
|
ИЛ-1ra sTNFR Декорин
|
|
Ингибиторы протеаз |
TIMP |
|
Ингибиторы комплемента |
DAF CR1 C1-ингибитор Фактор Н Витронектин |
|
Противовоспалительные цитокины
|
ИЛ-10 (15-23) ИЛ-4 ИЛ-13 TGF-β1[1] |
|
Противовоспалительные эйкозаноиды |
Простагландин E2, простагландин 12 (простациклин[2]) Липоксин А4 и В4 (24-36) |
|
Антитромботические молекулы |
Ингибитор тканевого фактора Активатор плазминогена тканевого типа Активатор плазминогена урокиназнного типа |
|
Внутриклеточная защита. |
|
|
Белки теплового шока (37-39) Гемоксигеназа-1 Антиоксиданты (СОД, каталаза, глутатионпероксидаза) Ингибиторы циклинкиназ (p21, p27)
|
|
[1] TGF-β группа цитокинов, с одной стороны обладающих противовоспалителным действием, а с другой стороны участвующих в процессах репарации тканей и приводящих, в частности, к активации процессов нефросклероза. Поэтому действие TGF-β на ткани почки может иметь как положительные, так и отрицательные стороны на различных этапах почечного континуума (32).
[2] Простациклин синтезируется эндотелием сосудов. Этот эйкозаноид во многом является антагонистом тромбоксана А2, так как снижает агрегацию тромбоцитов и вызывает вазодилатацию (40).
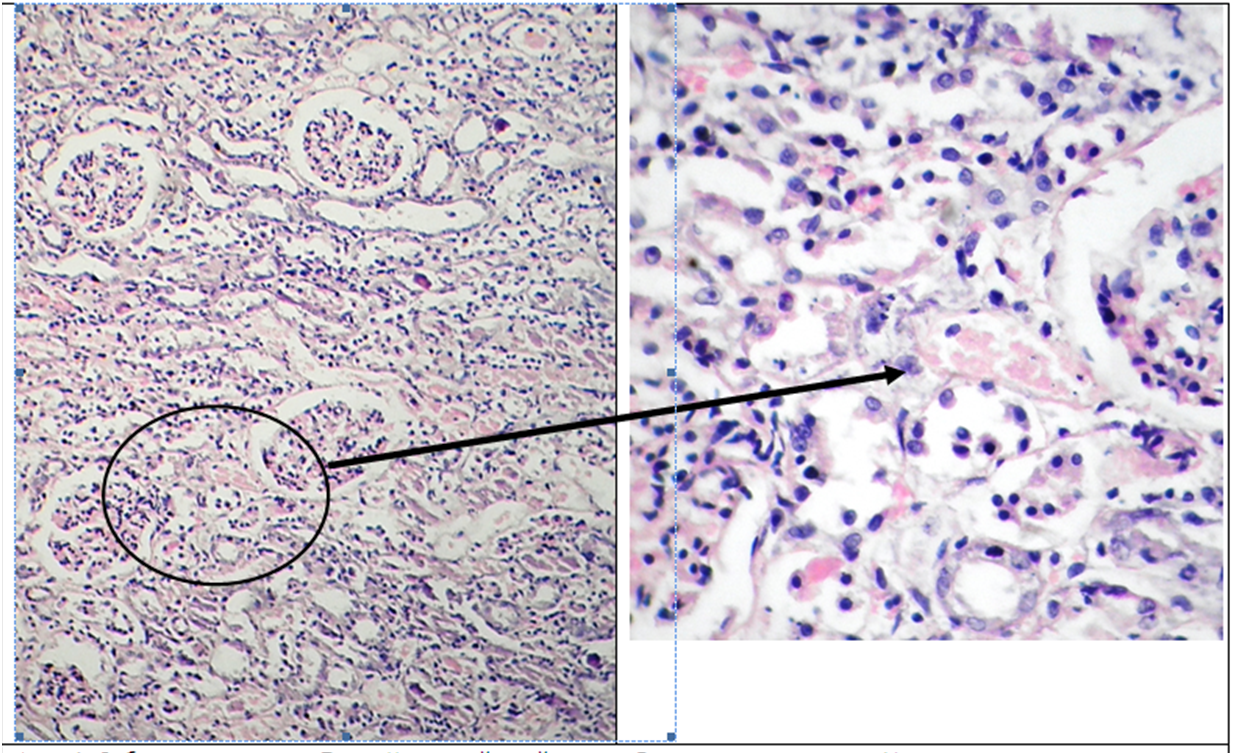
Фото 1. Собака, пекинес, м. 7 лет. Корковый слой почки. Лапчатость гломерул. Увеличение плотности и малокровие первичной микрокапиллярной сети. Расширение мочевого пространства клубочка. Дистрофия и атрофия высокого цилиндрического эпителия проксимпльных канальцев. Расширение и разрыв базальной мембраны канальцев. Скопление белковых депозитов в просвете канальцев. Гематоксилин-эозин. Ув.х400 слева и Ув.х1000 справа.
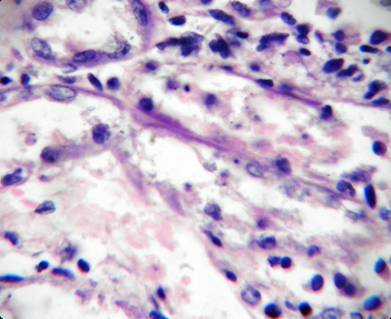
Фото 2. Собака, пекинес, м. 7 лет. Корковый слой почки. Дистрофия и атрофия высокого цилиндрического эпителия проксимальных канальцев. Расширение и разрыв базальной мембраны канальцев. Клеточно-белковые депозиты в просвете канальцев. Кариопикноз ядер эпителия канальцев. Гематоксилин-эозин. Ув.х1000.
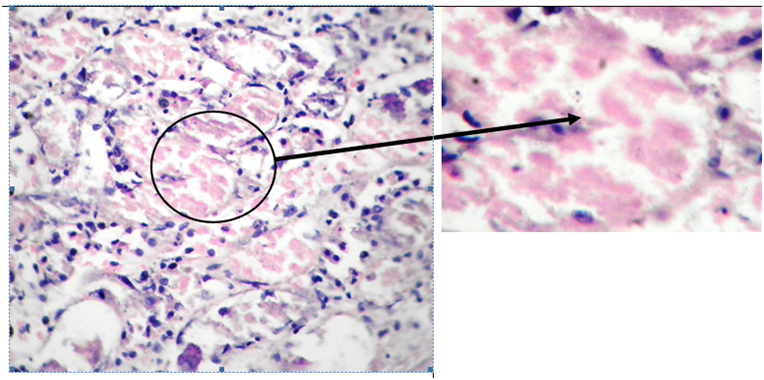
Фото 3. Собака, пекинес, м. 7 лет. Корковый слой почки. Дистрофия и атрофия высокого цилиндрического эпителия проксимальных канальцев. Расширение и разрыв базальной мембраны канальцев. Белковые депозиты в просвете канальцев. Кариопикноз ядер эпителия канальцев. Гематоксилин-эозин. Ув.х400 слева и Ув.х1000 справа.
Комментарии к фото 1,2,3.
Аминогликозиды: механизм токсического действия.
По медицинским данным нефротоксический ОКН (острый канальцевый некроз) диагностируется у каждого 10-го больного ОПН. Среди более чем 100 известных нефротоксинов одно из первых мест занимают лекарственные препараты, главным образом аминогликозидные антибиотики, применение которых в 10-15% случаев при¬водит к умеренной, а в 1-2% - к тяжелой ОПН.
Нефротоксичность аминогликозидов (канамицин, гентамицин, мономицин, неомицин (входит в состав ветеринарного препарата «Неопен»), тобрамицин) связана с наличием в их молекулах свободных аминогрупп в боковых цепях. Аминогликозиды в организме не метаболизируются, и 99% антибиотика в неизмененном виде выводится с мочой. Они фиксируются на апикальной мембране клеток проксимальных канальцев и петле Генле, связываются с везикулами, поглощаются путем пиноцитоза и секвестрируются в лизосомах канальцевого эпителия. При этом концентрация препарата в корковом веществе становится выше, чем в плазме. Для поражения почек аминогликозидами характерны увеличение в мембранах анионных фосфолипидов, в частности, фосфатидилинозитола, повреждение мембран митохондрий, сопровождающиеся потерей внутриклеточного калия и магния, нарушении окислительного фосфорилирования и дефицит энергии. Эти изменения приводят к некрозу канальцевого эпителия.
Также по собственным наблюдениям использование аминогликозидов в течении 5-10 дней приводит у кошек к стремительному накоплению большого числа агранулоцитов на границе коркового и мозгового слоев почки.
Фенистированный эндотелий как основной регулятор тонуса сосудов клубочка и важнейшее звено в патогенезе ГП и ГН.
Эндотелиальная дисфункция (прогрессирующее нарушение целостности фенистированного эндотелия, выстилающего капилляры клубочка), если и не всегда начальное, то в подавляющем числе случаев важнейшее звено в патофизиологии большинства ГП, ГН и вторичных тубулоинтерстициальных нефритов (ТИН) .
Фенистированный эндотелий капилляров почечного клубочка играет огромную роль в поддержании адекватного почечного кровотока, а значит и в функционировании почек как органа. Он синтезирует большое количество (БАВ), которые выполняют важную роль, как во многих физиологических процессах, так и при различных патологиях (гипериммунные реакции, процессы регенерации в фильтрационном барьере почки и т.д.). На поверхности клеток эндотелия находится большое количество рецепторов, воспринимающих сигналы как собственных БАВ, так и продуцируемых в других местах организма.
В физиологически нормальных условиях фенистрированный эндотелий активно препятствует процессам агрегации и коагуляции крови, а также спазмированию сосудов путем синтеза БАВ, таких как: оксид азота, простациклин, антитромбин III и др. Кроме того, эндотелий, образуя тромбомодулин, блокирует активные коагулянты, выделяющиеся печенью и находящиеся в плазме крови (тромбин). И, наконец, эндотелий адсорбирует антикоагулянты из плазмы крови, препятствуя адгезии и агрегации тромбоцитов на своей поверхности (гепарин, протеины С и S).
При повреждении эндотелий становится инициатором свертывания крови, сужения (спазма) сосудов и репарационных процессов в месте нарушения их целостности. Преобладание агрегантов и вазоконстрикторов объясняется при этом следующими основными причинами. Во-первых, повреждение или нарушение функции эндотелия подавляет секрецию антиагрегирующих, противосвертывающих и сосудорасширяющих веществ; во-вторых, эндотелий в этих условиях секретирует очень активные агреганты, коагулянты и вазоконстрикторы (7). В норме - это защитная реакция эндотелия сосудов, предохраняющая организм от потери крови. Но в микрокапилярной сети почек эти процессы репарации часто носят фатальный характер, поскольку являются начальным этапом гломеруло- и нефросклероза.
Уменьшение массы функционирующих нефронов, как универсальный фактор активации компенсаторных механизмов при нефропатиях.
Согласно теории В.М. Brenner уменьшение массы действующих нефронов и функционирующей почечной паренхимы, в не зависимости от первичных повреждающих факторов, приводит к активизации универсальных компенсационных процессов в функционирующих нефронах (гиперфильтрация) и инициирует необратимые процессы гломеруло- и нефросклероза. Эта теория была сформулирована после серии последовательных экспериментов, направленных на изучение механизмов повреждения остаточной почки здоровых животных после выполнения им субтотальной нефрэктомии (1-4). «Позже В.М. Brenner и соавт. установили, что эти морфологические изменения ассоциированы с нарушениями ауторегуляции внутрипочечной гемодинамики. Последние проявляются, в частности, снижением сопротивления преимущественно в афферентной артериоле, что приводит к возрастанию интрагломерулярного капиллярного плазмотока (гиперперфузия) и повышению внутриклубочкового гидравлического давления с увеличением скорости клубочковой фильтрации в каждом функционирующем нефроне (гиперфильтрация). Оказалось, что степень этих интрагломерулярных (внутриклубочковых) гемодинамических сдвигов коррелирует со степенью утраты почечной ткани. Так, если после удаления 50% массы действующих нефронов (МДН) клубочковая гиперфильтрация в действующих нефронах возрастает на 40-50%, то после удаления более 70% почечной ткани она возрастает более чем в два раза» (4).
Заключение.
Разнообразие повреждающих факторов при хронических асептических нефропатиях очень велико. Но векторы их воздействия в основном направлены на вазоконстрикцию артериол клубочка, что приводит к гиперфильтрации, и на процессы патологической (фатальной) репарации в поврежденных участках фильтрационного барьера. Именно это многообразие разнотипных, но однонаправленных эффекторных механизмов во многом и объясняет недостаточную эффективность даже комбинированной нефропротективной терапии (иАПФ, БРА ,кальциевые блокаторы, β-блокаторы), имеющей целью вазодилатацию эфферентных и/или эфферентных артериол, особенно на фоне прогрессирующего уменьшения массы форменных элементов почки вследствие гломеруло- и нефросклероза. Дальнейшая работа в области нефропротекции при хронических асептических нефропатиях лежит в плоскости исследования местных защитных механизмов почки, а также в изучении и внедрение в клиническую практику лекарственных препаратов, тормозящих различные этапы патогенеза путем активации почечных эндогенных систем самозащиты.
Литература.
1. Brenner B.M., Meyer T.W., Hosteller T.H. Dietary protein intake and the progressive nature of kidney disease: The role of hemody-namically mediated glomerular injury in the pathogenesis of progressive glomerular sclerosis in aging, renal ablation, and intrinsic renal disease. NEJM 1982; 307: 652-659.
2. Hostetter T.H., Olson J.L.,Rennke H. et al. Hyperfiltration in remnant nephrons. A potentially adverse response to renal ablation. Am TPhysiol 1981; 241: 85-93.
3. Olson J.L., Urdaneta A.G.,Heptinstall R.H. Glomerular hyalinosis and its relation to hyperfiltration. Lab Invest 1985; 42; 4: 387-398.
4. Томилина Н. А., Багдасарян А. Р. Механизмы нефросклероза и фармакологическая ингибиция внутри почечной ренин-ангиотензиновой системы как основа нефропротективной стратегии при хронических заболеваниях нативных почек и почечного трансплантата (Обзор литературы). Журнал "Нефрология и диализ" Т. 6, 2004 г., №3.
5. Böttinger EP, Bitzer M. TGF-beta signaling in renal disease. J Am Soc Nephrol. 2002 Oct;13(10):2600-10.
6. Sedor J.R., Nakazato Y., Konieczkowski M. Interleukin-1 and the mesangial cell. Kidney Int 1992;41:595–599.
7. Лупинская З.А. Эндотелий сосудов- основной регулятор местного кровотока. Вестник КРСУ / № 7, 2003 г.
8. http://en.wikipedia.org/wiki/Amyloidosis
9. Н.В. Чеботарева, И.Н. Бобкова, Л.В. Козловская, О.А. Ли. Значение нарушений механизмов самозащиты почки при хроническом гломерулонефрите. Журнал «Клиническая нефрология», 1-2011, стр. 8-14.
10. Visse R., Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function and biochemistry. Circ Res 2003; 92:827–839.
11. El Nahas A.M. Mechanisms of experimental and clinical renal scarring. Jn: Oxford Textbook of Clinical Nephrology ed. Davison, Cameron et al. 1998; 3: 1749-1776.
12. El Nahas A.M. Glomerulosclerosis: intrinsic and extrinsic pathways. Nephrol Dial Transplant 1996; 11: 773-777.
13. Wolf G.,Ziyadeh F.N., Thaiss F. et al. Angiotensin II stimulates expression of the chemokine RANTES in rat glomerular endothelial cells. J Clin Invest 1997; 100: 1047-1058.
14. http://en.wikipedia.org/wiki/Endothelin
15. Asadullah K., Sterry W., Volk H.D. Interleukin-10 therapy – review of a new approach. Pharmacol Rev 2003;55:241–269.
16. Fouquerbay B., Boutard V., Phillipe C. et al. Mesangial cell-derived interleukin-10 modulates mesangial cell response to lipipolysaccharide. Am J Pathol 1995; 147:176–182.
17. de Waal Malefyt R., Abrams J. et al. Interleukin-10 inhibits cytokine synthesis by human monocytes: An autoregulatory role of IL-10 produced by monocytes. J Exp Med 1991;174:1209–1220.
18. Olszina D.P., Pajkrt D., Lauw F.N. et al. Interleukin-10 inhibits the release of CC chemokines during human endotoxemia. J Infect Dis 2000;181:613–20.
19. Kuga S., Otsuka T., Niiro H. et al. Supression of superoxide anion production by interleukin-10 is accompanied by a downregulation of the genes for subunit proteins of NADPH oxidase. Exp Hematol 1996; 24:151–57.
20. Fouqueray B., Suberville S., Isaka Y. et al. Reduction of proteinuria in anti- glomerular basement membrane nephritis by interleukin-10 (IL-10) gene transfer. J Am Soc Nephrol 1996;7:1698–1701.
21. Tipping P.G., Kitching A.R., Huang X.R. et al. Immune modulation with interleukin-4 and interleukin-10 prevent crescent formation and glomerular injury in experimental glomerulonephritis. Eur J Immunol 1997;27:530–537.
22. Kitching A.R., Katerelos M., Mudge S.J. et al. Interleukin-10 inhibitsexperimental mesangial proliferative glomerulonephritis. Clin Exp Immunol 2002;128:36–43.
23. Papayianni A., Serhan C.N., Philips M.L. et al. Transcellular biosynthesis of lipoxin A4 during adhesion of platelets and neutrophils in experimental immune complex glomerulonephritis. Kidney Int 1995;47:1295–1302.
24. Claria J., Serhan C.N. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interaction. Proc Natl Acad Sci USA 1995;92:9475.
25. Conrad D.J., Kuhn H, Mulkins M. et al. Specific inflammatory cytokines regulate the expression of human monocyte 15-lipoxygenase. Proc Natl Acad Sci USA 1992;89:217–221.
26. Brinckmann R., Topp M.S., Zalan I. et al. Regulation of 15-lipoxygenase expression in lung epithelial cells by interleukin-4. Biochem J 1996; 318:305–312.
27. McMahon B., Mitchell S., Brady H.R. et al. Lipoxins: Revelation on resolution. Trends Pharmacol Sci 2001;8:391–395.
28. Colgan S.P., Serhan C.N., Parcos C.A. et al. LipoxinA4 modulates transmigration of human neutrophils across intestinal epithelial monolaeyrs. J Clin Invest 1993; 92:75–82.
29. Ohse T., Ota T., Kieran N. et al. Modulation of interferon-induced genes by lipoxin analogue in anti-glomerular basement membrane nephritis. J Am Soc Nephrol 2004;15:919–927.
30. Godson C., Mitchell S., Harvey K. et al. Cutting edge: Lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol 2000;164:1663–1667.
31. McMahon B., Mitchell D., Shattock R. et al. Lipoxin, leukotriene and PDGF receptors cross-talk to regulate mesangial cell proliferation. FASEB J 2002;16:1817–1819.
32. Fierro I.M., Kutok J.L., Serhan C.N. Novel lipid mediator regulators of endothelial cell proliferation and migration: Aspirin-triggered-15R-lipoxin A4 and lipoxin A4. J Pharmacol Exp Ther 2002;300:385–392.
33. Katoh T., Takahashi K., DeBoer D.K. et al. Renal hemodynamic actions of lipoxins in rats: A comparative physiological study. Am J Physiol 1992; 263:436–442.
34. Wu S.-H., Liao P.-Y., Yin P.-L. et al. Elevated expression of 15-lipoxigenaseand lipoxin A4 in children with acyte poststreptococcal glomerulonephritis. J Pathol 2009;174:115–122.
35. Boutet P., Bureau F., Dengand G. et al. Inbalance between lipoxin A4 and leukotriene B4 in chronic mastitis-affected cows. J Dairy Sci 2003; 86:3430–3439.
36. Wu S.-H., Liao P.-Y., Yin P.-L. et al. Inverse temporal changes of lipoxin A4 and leukitrienes in children with Henoch-Schonlein purpura. Prostaglandins, Leukotrienes and essential fatty acids 2009;80(4):177–183.
37. Olszina D.P., Pajkrt D., Lauw F.N. et al. Interleukin-10 inhibits the release of CC chemokines during human endotoxemia. J Infect Dis 2000;181:613–20.
38. Aufricht C., Lu E., Thulin G. et al. ATP releases HSP72 from protein aggregates after renal ischemia. Am J Physiol Renal Physiol 1998;274:268–274.
39. Jorgensen C., Gedon E., Jaquet C. et al. Gastric administration of recombinant 65kDa heat shock protein delays the severi of type II collagen induced arthritis in mice. J Rheumatol 1998;25:763–767.
40. http://en.wikipedia.org/wiki/Prostacyclin
41. Фундаментальная и клиническая физиология: Учебник для студ. высш. учеб. заведений / Под ред. А. Г. Камкина и А. А. Каменского. — М.: Издатель¬ский центр «Академия», 2004. — 1072 с., стр. 993-994

|
WWW.VETNEFRO.RU Научно-практическая ассоциация ветеринарных нефрологов и урологов
© Все права защищены 2024г. Использование любых материалов сайта - запрещено!
|